Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy: from ablation to heart transplantation.
Olga Blagova, Alexander Nedostup, Dmitry Shumakov, Vitaly Poptsov, Anna Shestak, Elena Zaklyasminskaya
We present 38-years male patient. He has suffered from muscle weakness since 5 years. Arrhythmias appeared at the age of 32. In 37 years he had sick sinus syndrome, transient AV block II degree, paroxysmal atrial fibrillation, atrial flutter, ventricular arrhythmias. At this time, dilated cardiomyopathy was also detected. The evaluation revealed knees and elbows contractures, increased level of creatine kinase. The genetic testing revealed a frame shift deletion c.del619C in the emerin (EMD) gene and c.IVS4-13T> A in the lamin (LMNA) gene, and c.del619C deletion in the heterozygous state in a patient`s mother. Radiofrequency ablation of cavotricuspid isthmus, implantable cardioverter-defibrillator (ICD) implantation were performed. Heart transplantation was performed nine months later, due to severe heart failure and electrical storm. A morphological evaluation revealed sclerosis, atrophy and hypertrophy of cardiomyocytes. He underwent an induction therapy with (basiliximab) methylprednisolone, tacrolimus, mycophenolate after heart transplantation. During 40 months after transplantation, patient`s condition is satisfactory.
Conclusion: heart failure in Emery-Dreifuss muscular dystrophy can progress quickly unless the previously stable condition. The use of correct regimens of immunosuppression therapy provides good long-term results of the heart transplantation
Correspondence to: Blagova Olga, Sechenov First Moscow State Medical University 119992, Moscow.
If we detect arrhythmias and dilated cardiomyopathy (DCM) in young patients with symptoms of myopathy and / or increased levels of total creatine kinase, we should always exclude a hereditary neuromuscular disease. Genetic testing in DCM with the neuromuscular disease gives positive answers much frequently (62%) than in family cases (25%) and sporadic (8%) forms of DCM [1]. There are large varieties of clinical forms of progressive myopathy with heart failure. There is a wide variety of progressive myopathy involving heart failure. They are different in clinical forms, type of inheritance, age of debut and prognosis. The aim of the report is to discuss some common questions of diagnostics and treatment of cardiomyopathy in muscular dystrophy illustrated by the case of our patient.
A male patient of thirty-eight years came to our clinic in May 2012 with moderate weakness in the proximal muscles of the limbs, presyncope episodes not associated with physical activity, proximal muscular weakness, dyspnea at moderate physical activity and episodes of palpitation.
Patient`s mother was implanted pacemaker at the age of fifty-four [Fig. 1], his sixty-six years old father had a stroke. Two patient`s sons 3 and 11 years were clinically healthy. The patient has smoked since a young age. Otherwise he had a healthy lifestyle.
Figure 1. The pedigree of patient.
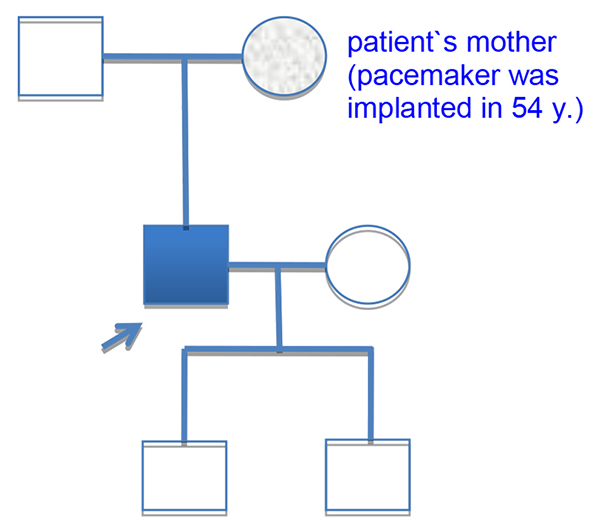
Proband is indicated by a blue square. His mother was implanted pacemaker in 54 years. The nature of her disease was unknown. Two patient`s children are clinically healthy.
The patient has suffered from low progressive skeletal myopathy since childhood. Since 5 years he have had progressive muscular weakness, frequent episodes of falling. In the age of six he was diagnosed muscular dystrophy. He has had arrhythmias and minimal ejection fraction (EF) reduction since the age of 32. Palpitatons and presyncope appeared and increased in 2012. Echocardiography and Holter monitoring showed signs of DCM, sick sinus syndrome, transient AV block II degree type 1, paroxismal atrial flutter and fibrillation, more than 4000 premature ventricular beats (PVBs) and unsustained ventricular tachycardia. However he had normal coronary angiograms.
Patient`s height is 180 cm, weight 77 kg. He had gait disorders, moderate knees and elbows contractures. He had no edema. His respiratory rate was 18 per minute, and there were no wheezing in lungs. Heart rate was 56 per minute, premature beats 2-4 per minute, it was no cardiac murmur, blood pressure was 110/70 Hg mm, no ascites and hepatomegaly. We suspected Emery-Dreifuss muscular dystrophy (EDMD) cause of combination of muscle weakness, high level of creatine kinase (576 U/l), normal intellect, DCM and ventricular and supraventricular arrhythmias. Direct Sanger sequencing was in progress.
However, the choice of treatment strategy was not easy because we had no definite diagnosis. We decided to do a biopsy of the myocardium or skeletal muscle, radiofrequency ablation of supraventricular arrhythmias, pacemaker implantation. However, given the genetic nature of the DCM and its progressive course was decided to implantable cardioverter-defibrillator (ICD) implantation.
He was undergone radiofrequency ablation of cavotricuspid isthmus in Bakoulev Center. The dual-chamber ICD implantation was also performed. Amiodarone showed good clinical effect. Left ventricular (LV) EF was forty-three percent. However, he got worse four months later. The patient had palpitations, progressive dyspnea and edema. Atrial flutter, low LV EF (less than twenty percent) and severe mitral and tricuspid regurgitation were detected at this time.
Emery-Dreifuss muscular dystrophy diagnosis have been already genetically confirmed by that time. They found two genetic variants [Fig. 2]:frame-shift deletion c.del619C in emerin (EMD) gene causing premature stop-codon appearance and protein shortening (p.236X); 2) intron replacement c.IVS4-13T>A in lamin (LMNA) gene with unknown clinical significance. Both variants were not found in control group of 100 healthy volunteers.
Figure 2. Results of DNA diagnostics.
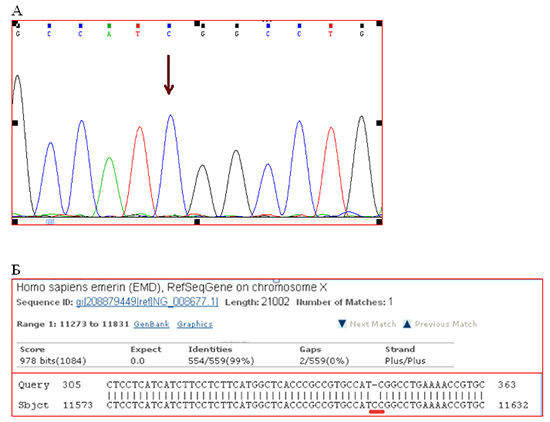
A. Detail of direct Sanger sequencing of exon 6 EDM gene. The arrow indicates place of the lost nucleotide C. B. Compare fragments of the nucleotide sequence of exon 6 of the gene patient EDM («Query») with the reference sequence of exon 6 of the gene EDM patient NG_008677.1 («Sbjct"). Red line underlined the place of deletion.
The patient was in our clinic in February 2013. He had tachycardia 120 beats per minute, irregular pulse, deficits 10-15 beats per minute and severe congestive heart failure symptoms with signs of hepatomegaly and cholestasis. The level of creatine kinase remained high (458 U/l). His thyroid status showed euthyroid hyperthyroxinemia.
We had to exclude myocarditis because of heart failure dramatic progression in previously stable patient. Real-time polymerase chain reaction found no viral genome in blood. The level of anti-heart antibodies moderately increased (1:160 toward endothelial antigens and antigens of conductive system). By the way, it could be secondary immune reaction of cardiomyocytes’ damage.
ECG showed [Fig. 3A] atypical atrial flutter with FF waves period 0,20 s., heart rate was 90/min, right bundle brunch block (QRS duration 0.16 s.), both ventricles hypertrophy signs. Holter monitoring revealed sustained atrial flutter with moderate tachycardia, atrial flutter (2:1, 3:1, 4:1), ICD VVI pacing (20% of QRS) 75 beats per minute, average heart rate was 85/minute daytime and 76/minute nighttime, 787 PVBs, 18 couplets, 1 triplet. Left ventricular ejection fraction was about thirty percent, though left ventricular end-diastolic diameter (7.0 cm) and volume (305 ml) and left and right atria volume (187 ml and 148 ml) were significantly dilated. The patient also had moderate pulmonary hypertension (46 mm Hg) and tricuspid regurgitation (II degree). Multi-slice computed tomography showed no evidence of coronary atherosclerosis and cardiac thrombosis.
Figure 3. Electrocardiogram.
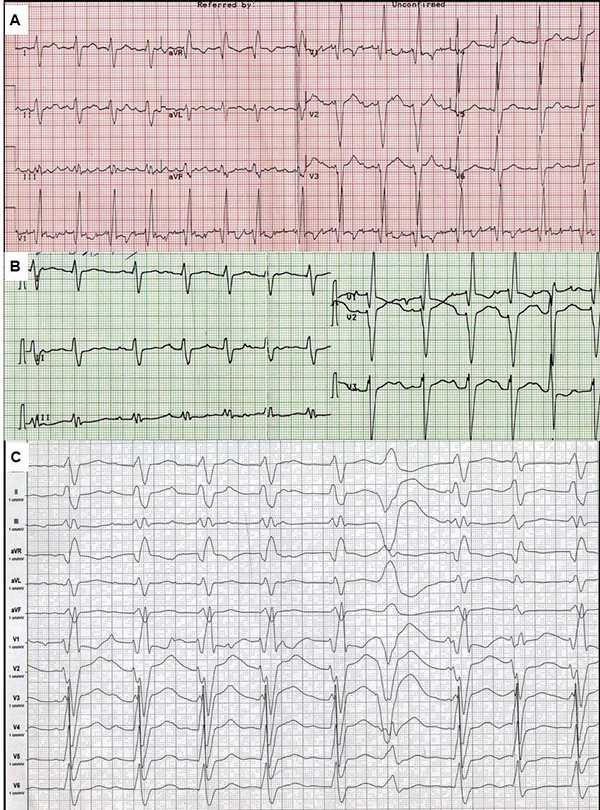
Speed 25 mm/s (A, B), and 50 mm/s (C). A - ECG in February 2013, B - ECG after electrical cardioversion. C - ECG after repeated ICD shocks.
There were several possible causes of deterioration in the condition. It could have been the increasing of tricuspid regurgitation and ICD implantation asynchrony, relapse of sustained tachyarrhythmia and the development of disease. Thus, we discussed CRTD reimplantation and heart transplantation. Electrical cardioversion was successful [Fig. 3B]. It was complicated by sustained ventricular tachycardia and did not significantly improve the condition. He was treated with perindopril 2.5 mg, amiodarone 400 mg, warfarin 2.5-3.75 mg and furosemide 40-60 mg per day. Despite of the amiodarone treatment he had an electrical storm (ventricular tachycardia and fibrillation) with multiple ICD shocks. ECG after shocks is shown in [Fig. 3C].
He was urgent hospitalized to Shumakov Federal Research Center of transplantology and artificial organs. He was implanted extracorporeal membrane oxygenation (ECMO) system. He was undergone successful orthotopic heart transplantation. ECMO system was removed the next day after heart transplantation. Basiliximab induction therapy, prednisone, tacrolimus and mycophenolate were assigned later. Temporary pacing 90-100 per minute have been supported for 4 weeks. There were no signs of rejection in the control myocardium biopsy (0-I degree).
Morphology of the explanted heart showed 470 g weight, 11х9х4.5 cm size and normal coronary arteries. The myocardium was flabby, homogeneous, pink-brown. Histology of the explanted heart showed diffuse myocardial fibrosis, cardiomyocytes polymorphism (atrophy and hypertrophy), perinuclear edema, decaying nuclei (apoptosis) [Fig. 4]. There were no signs of associated myocarditis.
Figure 4. Morphology of explanted heart (left ventricle).
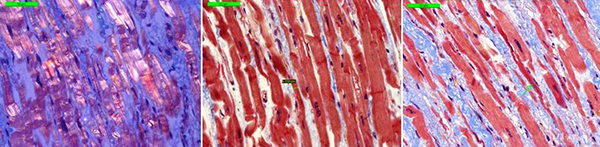
Microscope 40x magnification. Left picture (polarized light) shows thin fibers, interstitial sclerosis. The central and the right picture (Masson's Trichrome stain) showed thin cardiomyocytes in the longitudinal sections; interstitial sclerosis, thin fibers, nuclear decay).
Forty-month follow-up showed significant health improvement. There were no signs of rejection and other specific complications, muscle weakness did not progress. Patient took up to work and he had a great physical activity.
We also found the heterozygous state deletion c.del619C in gene of emerin in the 63-years-old patient`s mother. She has been diagnosed coronary heart disease (without signs of atherosclerosis) and arterial hypertension for a long time. She had satisfactory functional status. She was implanted pacemaker because of sick sinus syndrome, AV block with syncope. Manifestation of X-linked Emery-Dreifuss muscular dystrophy included moderate DCM (LV end-diastolic diameter 6.4 cm, EF about 50%). We did not need any genetic testing of patient's sons because of X-linked recessive type of disease.
As we know, there are three major forms of muscular dystrophy, which involve heart: Duchenne, Becker and Emery-Dreifuss. Degrees of the involving are variable.[2]. We suspected EDMD in our patient because he had a combination of gait disorders, moderate knees and elbows contractures, high levels of creatine kinase, normal intellect, DCM-form heart involvement and AV block. Genetic test confirmed the diagnosis
Genetic nature of Emery-Dreifuss muscular dystrophy (EDMD) was described in 1994, when mutations were identified in the gene emerin [3]. There are at least five other genes which are responsible for the development of the disease (LMNA, SYNE1, SYNE2, TMEM43 and FHL1). The combinations of mutations in two genes could be reason for quick progression of cardiomyopathy [4]. Other reason could be production of anti-heart antibodies, for example to troponin I [5]. In addition, intervention (radiofrequency ablation and ICD implantation) could disrupt compensatory mechanisms.
There are no guidelines for the treatment of EDMD patients. The optimal treatment strategy, especially surgical, should be individual. There are just a few reports of radiofrequency ablation in patients with myopathies [6]. The reduction of arrhythmia does not improve the disease in case of severe cardiomyopathy. The use of an ICD could be considered in EDMD patients if there are indication for pacing and evidence of ventricular arrhythmias (class IIb, level B), [7]. We believe that implantation of ICDs was useful to our patient and gave him a chance to survive up to heart transplantation.
However, ICD implantation could have been one of the causes of arrhythmias and heart failure progression. Artificial interventricular dyssynchrony could be the reason of deterioration. Our patient did not have a high right ventricle stimulation (20%). The restoration of sinus rhythm did not improve the condition. Development of electric storm determined the further tactic. It is not typical for patients with MDMD. Heart failure is more common reason for heart transplantation. Italian register of LMNA-associated myopathies
includes 78 patients, 17 (22%) have EDMD2, ICD or pacemaker was implanted in 41 (53%) and heart transplantation was performed in 8 (10.3%) myopathic patients [8]. There were two percent of patients with muscular dystrophies through all cases of heart transplantation [9].
Surgeons usually face some problems in transplantation connected with myopathy. There are higher perioperative risk in general, difficulty of anesthesia due to respiratory and neck muscles damage, higher risk of aspiration, rhabdomyolysis, malignant hyperthermia metabolism, worsening of peripheral myopathy due to steroids. Total IV anesthesia and non-depolarizing muscle relaxants are the optimal for use. The same was used in our patient.
The immunosuppression therapy was effective and relative safety in our patient. He got basiliximab (monoclonal antibodies to interleukin-2 receptor, CD25), tacrolimus and mycophenolate. The recent study estimated 10 years’ experience this scheme compared with cyclosporine: at equally high survival rate (66.7% and 80.0%), tacrolimus provided a lower incidence of acute rejection and vasculopathy transplanted heart [10].
Conclusion. Cardiomyopathy in patients with primary myopathy (Emery-Dreifuss muscular dystrophy, EDMD) could develop quickly despite of early stable course. These patients should be regular evaluated by cardiologist. Two genes mutation could explain severe cardiomyopathy in our patient with EDMD. Myocarditis should be excluded in all cases of «unexplained» decompensation in EDMD patients. Verification of genetic myopathy variant with cardiac involvement is useful to decide the way of treatment, including surgery. Indications for radiofrequency ablation and ICD implantation in EDMD patients should be choose in case of immediate and long-term prognosis. Although patients with EDMD have peripheral myopathy and limitations of anesthetics use, heart transplantation could be successful done if we use correct regimens of immunosuppression therapy. Women with X-linked EDMD have a mild form of disease. It could looks like more frequent heart diseases so it could not be diagnosed for a long time.
Authors of the report have no conflicts of interest.






