Sinus Node Dysfunction in Atrial Fibrillation: Cause or Effect?
Anna Kezerashvili, MD1, Andrew K. Krumerman, MD, John D. Fisher, MD
1From the Department of Medicine, Cardiology Division, Arrhythmia Service, Montefiore Medical Center and the Albert Einstein College of Medicine.
Key Words : Atrial Fibrillation, Sick Sinus Syndrome, Electrical Remodeling.
Correspondence to: Dr. John D. Fisher, MD,Montefiore Hospital,Cardiology N-2, Arrhythmia Offices,111 E. 210th Street,Bronx, NY, 10467, USA
A connection between Sick Sinus Syndrome (SSS) and atrial fibrillation (AF) has been recognized in the literature as early as 1960 and is well described by Irené Ferrer1 in her early classic book on SSS. In this article we will review the evidence supporting AF inducing SND. In addition we will explore the evidence supporting the notion that SND causes and promotes the development of AF.
The term SSS was coined by Ferrer in 1968 who grouped together all the anatomic etiologies of sinus node disease (SND).1Table 1 . We will explore how some patients with underlying SND can have coexisting AF either the effect of SND on atria via various mechanisms or via the underlying disease of the atria. SND is frequently associated with AF, forming the basis of the ”tachycardia-bradycardia syndrome.”2
Table 1. Some Causes of Sinus Node Dysfunction.
| Underlying Heart Disease |
Other Predisposing Factors |
Reversible Causes |
| Coronary artery disease |
Friedreich’s ataxia |
Atrial Fibrillation |
| Ischemic sino-atrial disease |
Muscular Dystrophy |
Atrial Flutter |
| Rheumatic heart disease |
Collagen Disease |
Acute Ischemia |
| Dilated cardiomyopathy |
Amyloidosis |
Pericarditis |
| Restrictive cardiomyopathy |
Hemochromatosis |
Myocarditis |
| Hypertrophic cardiomyopathy |
Familial sinoatrial disease
Muscular Dystrophy |
Pneumonia |
The mechanisms that give rise to AF, whether paroxysmal or chronic have been investigated by many, yet there is still no consensus on many of the physiologic, structural, mechanical or electrical etiologies of AF. Disease of the SN has been implicated as one of the many underlying causes of AF. Table 2 . The conceptual framework for understanding AF mechanisms has its groundwork in ideas developed in early twentieth century.3 The concept of the “wavelength of re-entry” developed by Allessie et al emphasized the notion of the size of the wavelength on functional re-entry in his “leading circle” hypothesis.4 This work experimentally confirmed the hypothesis of Moe et al. of the role of multiple re-entrant wavelets in the perpetuation of AF in animal models.5 Other theories such as “focal source hypothesis” and “sustained re-entry”6,7 have also been validated. A role for pulmonary veins drew attention when Haissaguerre et al. reported that rapid activations in the pulmonary veins may be triggering AF.8 Electrical remodeling is believed to be a central point in perpetuation of AF.
Table 2. Causes of Atrial Fibrillation.
| Underlying Heart Disease |
Other Predisposing Factors |
Reversible Causes |
| Coronary artery disease |
Hyperthyroidism |
ETOH intake |
| Valvular rheumatic disease |
Genetic/Familial |
Electrocution |
| Hypertensive heart disease |
Bronchopulmonary disease |
Surgery |
| Dilated cardiomyopathy |
Diabetes |
Myocardial Infarction |
| Hypertrophic cardiomyopathy |
Congestive Heart Failure |
Pericarditis |
| Restrictive cardiomyopathy |
Familial sinoatrial disease
Muscular Dystrophy
|
Pneumonia |
| Atrial ischemia |
Increased left atrial size |
Myocarditis |
| None (“Lone AF”) |
Autonomic nervous system |
Drug-electrolyte abnormalities |
Our knowledge of the formation of AF is far from complete, but work by Hudson and Laippestad in the early 1960’s suggested that damage to the sino-atrial
(SA) node was an important factor.9,10 When the sinus impulse formation is depressed in the presence of SND, an atrial extrasystole may occur during the slow atrial cycle. Most atrial extrasystoles are followed by a compensatory pause.If there is SND, the pause may be prolonged, allowing other atrial “escape” foci to fire. As stated by Ferrer: “If a dominant pacemaker ceases its activity, or slows its rate below that of a lower pacemaker, the latter will escape and control the cardiac rhythm.”1 In a substrate such as dilated or ischemic atria, any slowing of the SN, either via compensatory pause from extrasystoles or failure of sinus impulse formation, will allow sufficient time for multiple atrial foci to mature and fire. Since the foci are coming from an irritated atrial tissue, they may form frequent, multifocal, or “chaotic” atrial rhythms that frequently herald AF.11 Early works by Killip and Bennet have shown that in some patients atrial extrasystoles are critical to the initiation of AF and AFL.12,13
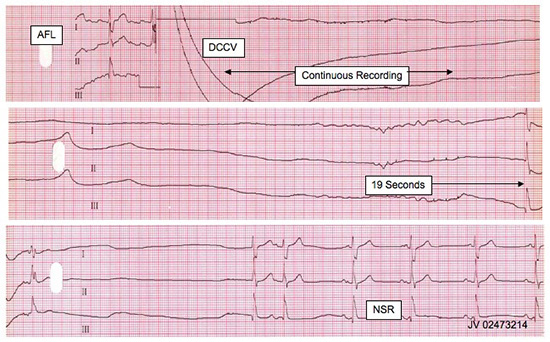
Figure 1. Prolonged asystole following cardioversion, followed by resumption of sinus rhythm. AFL: atrial flutter. DCCV: direct current cardioversion. NSR: normal sinus rhythm.
SND also may facilitate re-entry. In an atrium with SND early premature impulses originate from areas other than SN. Spach demonstrated regional differences of repolarization in the atrium.14 He noted that the duration of the longest atrial action potential (ADP) occurs in the area of the SN, with stepwise decrease in ADP as the distance from SN increases. Thus, impulses originating from sites other than the SN lose the protective effect of the long action potential and may result in conduction block and re-entry.14 Whether from a single circuit or multiple circuits, re-entry has been a dominant conceptual model of AF.3
Re-entry is also facilitated by an increase in the dispersion of excitability recovery during sinus bradycardia as reported by Han.15 According to Han, AF results from non-uniform recovery of excitability.16 AF may occur whenever there is dispersion of atrial refractoriness. Bradycardia further increases the dispersion of canine atrial refractoriness.15 A study by Luck et al. confirms that prolonged non-uniform refractoriness and increased dispersion of refractoriness are features of SND in humans.17 Unfortunately, these features persist even during atrial pacing, reflecting intrinsic atrial disease and possibly explaining why atrial pacing is relatively ineffective at preventing AF.17
Ischemic damage to the SA node alone, with no other atrial wall disturbances such as fibrosis, stretch or muscle loss may result in chronic atrial fibrillation (CAF). In a pathologic analysis of hearts with chronic and short-term AF, Davies noted that stenosis in the sinus nodal artery was common in patients with CAF.18 In a study of post CABG patients with AF and sinus rhythm (SR) angiograms showed SA nodal artery disease in 9 out of 25.19
Overall, a combination of atrial extrasystoles, shorter atrial ADP, dispersion of excitability recovery and ischemia to the SN itself are mechanisms by which SND may cause and promote AF.
Many have speculated on the concept that AF itself may alter a normal SN or promote preexisting SND. The concept of one pacemaker dominating the cardiac rhythm of the atria comes into play where AF is believed to shut down the SN by long term overdrive suppression of its activity. Hocini et al. found that patients with PAF and prolonged sinus pauses (>3sec), had improved SN function after ablation. Sinus pauses were thought to be due to long-term suppression of SN activity.20 Turitto reported a series of patients that demonstrated atrial standstill for up to 12 hours following ablation of long-standing AFL and AF.21 These findings suggest functional depression of sino-atrial activity. In 2/3 patients, depression was permanent and a pacemaker was needed. In contrast, Palma et al. noted to return to normal sinus function one day following ablation of chronic (25 years) atrial flutter.22 Prolonged suppression of sino-atrial activity can be attributed to both molecular and cellular mechanisms. Electrical remodeling and AF induced changes in atrial substrate work in concert to alter SN function.
Atrial Electrical Remodeling
Electrical remodeling can be largely attributed to intracytoplasmic calcium overload within atrial myocytes.3 Upon initiation of AF, intracellular calcium increases and results in subsequent down-regulation of inward calcium channel current and calcium channel alpha subunit production. Reduced inward calcium current during this electrical remodeling of the atrium also reduces SN impulse formation.23,24 Ischemia and calcium: Many studies have shown that AF can produce transient myocardial ischemia. In a study by Knoll et al. on myocyte nuclei of ischemic hearts, increased transcriptional activity for sarcoplasmic Ca2+ ATPase, resulted in decreased cytoplasmic Ca2+ concentration, which in turn shortened the ADP.25
3 had partial recovery, and 3 had no recovery of diaphragmatic function.
SAF results in a tenfold increase in atrial rate, initially increasing cellular Ca2+ loading.24 This threatens cell viability which triggers down regulation of the ICa channel, ultimately decreasing cytoplasmic Ca2+ concentration thereby promoting AF by decreasing the atrial effective refractory period (AERP) and negatively affecting SN impulse formation.3 Marked reductions in the densities of ICa channel, transient outward K+ current, (Ito) and ultra rapid delayed rectifier K+ current (Ikur) in atrial myocytes from patients in CAF were found in a study by Wagoner.26 These changes appear to be caused by a reduction in mRNA concentration of the respective channel alpha-subunits27 and reduction of L-type Ca2+ channel, SR, Ca2+ ATPase mRNA levels in patients with established AF.28
BSN electrical remodeling was demonstrated in a dog model using chronic pacing to maintain AF by Elvan and Zipes.29 In this model, 15 dogs were rapidly paced either in the ventricle or atrium for 2-6 weeks. As compared to controls, dogs with rapid atrial pacing were noted to have prolongation of P wave duration and sinus node recovery times (SNRT), indicating slowed conduction and depressed SN activity. In addition, shortening of AERP and reduced intrinsic heart rate were noted. These changes in the SN promote AF.Further, the duration of inducible AF was increased with atrial pacing compared with control dogs, and those paced only in the ventricle. Perhaps unexpectedly, the duration of AERPs became independent of basic drive cycle length. These findings suggest the existence of atrial memory in these models. We will revisit this concept later in this review. Suppression of SN function following initiation and maintenance of AF supports the notion that AF begets SND.
There is a wealth of evidence supporting the idea that AF temporarily or permanently affects SN activity.On a short term basis, prolongation of SNRT has been demonstrated following overdrive atrial pacing and has been attributed to the interaction between sino-atrial conduction and recovery of automaticity.30,31 Hadian et al. demonstrated SNRT prolongation occurring with 10-15 minutes of rapid atrial pacing, consistent with early SN electrical remodeling.32 Patients with CAF who undergo cardioversion, have an increased prevalence of SND. Such patients were noted to have prolonged SNRTs and delayed intra-atrial conduction times, showing that prevalence of at least temporary sinus nodal dysfunction was high in these patients.33 In addition, early recurrence of AF is frequently observed.34
AF is also associated with significant atrial structural changes.Atrial enlargement in CAF can cause structural changes to the SN affecting its activity. Increased atrial size combined with rapid heart rates predispose to atrial ischemia and further SND.23,35,36 Rapid ventricular rates during periods of AF may cause transient hemodynamic changes, in turn affecting sympathetic tone. Elvan and Zipes suggest that since AF leads to an increased sympathetic tone ,29 it in turn affects SN function, since autonomic tone is a major factor in regulation of sino-atrial conduction and SN automaticity.37,38,39 Ischemia, stretch, increased atrial mass and changes in autonomic tone are associated with interstitial fibrosis, connexin disarray, cellular damage and apoptosis of atrial tissue.23,36 Overall, AF can alter the structure and function of the SN via overdrive suppression, ion channel remodeling, cellular remodeling, transient myocardial ischemia, atrial enlargement and increased sympathetic tone.
CAF and PAF Affect Sinus Nodal Function Differently
Electrical remodeling of the atrium and SN may result from prolonged changes in atrial rate.23 The “Four Time Domains in Adaptation to Heart Rate” proposed by Alessie represent the different adaptative processes by the atria in response to changes in heart rate.4 (Table 3). This simplified scheme describes various levels or remodeling that occur between short term (metabolic- i.e. ion concentrations) and very-long-term (years – i.e. anatomical remodeling, fibrosis and muscle loss) changes to atrial substrate. These time domains help to elucidate pathophysiological differences between atria with PAF and CAF.
Table 3. Four Time Domains*.
| Short term changes – metabolic |
Increased/decreased ion concentrations |
| (lasts seconds – minutes) |
Increased/decreased ion pump activity |
| Moderate-term changes expression
(via electrical remodeling synthesis,
hours to days) |
Changes in ion channel gene |
|
Increased/decreased protein |
|
assembly |
| Long term –contractile Myocardium remodeling
(weeks) |
Hibernating myocardium (reversible damage) |
| Very long term – anatomical Remodeling (months-years)
|
Structural damage to myocardium (irreversible damage mostly) (scarring, fibrosis, fatty infiltration) |
Alessie prefers to avoid the term “remodeling” related to short-lived metabolic or electrical changes, and reserves “remodeling” for changes that occur after a period of time when there are changes to structure, cells and ion channels.4 Others use the term “electrical remodeling” for changes that do not (yet) include structural changes.25,32,40,41,42
Autopsy studies have revealed significant changes to atrial structure in patients with CAF consistent with those described in Alessie’s fourth time domain. In a study by Davies, where the hearts of 100 deceased patients with AF were examined, clear pathologic differences between chronic and short-term AF were found.45 Out of 74 patients with chronic AF 54 had SA node muscle loss. This was in contrast to patients with short-term AF where no muscle loss or damage to the SN, was noted.46 Thus, in chronic AF, remodeling of the SN occurs at many different levels; altered ion channel gene expression,25,40 irreversible structural damage via atrial stretch with increased atrial volume,35,43,44 and direct damage to the SN via increased metabolic demand accompanied by reduced coronary reserve to the SA nodal artery.36
Paroxysmal AF has also been implicated as a cause of SN electrical remodeling. In a study by Hadian et al., short-term rapid atrial pacing significantly prolonged sino-atrial conduction time (SACT) and corrected SNRT.32 A study by Goette et al demonstrated decreased AERP within 30 minutes of rapid pacing without atrial stretch.41 AF can decrease AERP after ten minutes.47 Decreases in AERP promote AF by decreasing the reentrant circuit wavelength (wavelength =AERP x CV (conduction velocity)), allowing the atria to accommodate a larger number of functional re-entry circuits.3 These findings were confirmed by Yue et al who noted a decrease in L-type Ca² current and Ca²-independent transient outward current (Ito) in the atria during rapid atrial pacing.48 Such short term changes in outward calcium current decrease atrial refractoriness and further promote AF.Dynamic decreases in atrial refractoriness were observed in all patients with PAF suggesting that electrical remodeling developed regardless of baseline AERPs.42 These short term changes fall into Time Domains 1 and 2, and may be reversible over a short period of time.
Atrial Memory And Short Term Atrial Electrical Remodeling
Memory has been described as a “specialized form of remodeling…induced by a preceding period of altered electrical activation.”49 One would then expect AF or rapid atrial pacing to induce atrial memory effects. In a study by Morillo, sustained AF could not be induced by electrical stimulation in dogs at baseline.35 After rapid pacing (400 bpm) for 6 weeks, AF was readily inducible by 1-3 extrastimuli.These findings suggest that atrial memory or atrial electrical remodeling predispose to AF.35 Duration of inducible AF was increased in paced dogs, while AERPs became independent of the basic drive cycle length.29 Left atrial (LA) pacing in mongrel dogs was associated with a decreased atrial gradient (the root mean square of three P-Ta voltage time integrals) and with occurrence of atrial tachycardias during pacing, that persisted during recovery from pacing.50 This suggests that altering the atrial activation sequence induces atrial memory, without altering the AERP. In another study by Herweg et al. displacement of the atrial gradient vector occurred during recovery from LA pacing, was more marked at rapid pacing rates, and manifested accumulation and resolution consistent with atrial cardiac memory.51 Changes observed during recovery from LA pacing appeared to affect repolarization primarily and were expressed electrographically as an altered P-Ta voltage-time integral.51 As stated by Goyal, changes in repolarizing currents and AP durations are elements of atrial memory.52 After electrical or drug cardioversion, AF vulnerability remains high, with single premature beats re-inducing drug-cardioverted AF and frequent recurrence for 1-2 days after electrical cardioversion.53,54 These observations of AF recurrence after cardioversion or short term pacing of the LA are all suggestive of atrial memory. Still, others doubt that atrial memory has been demonstrated, looking at AERP changes rather than P-Ta voltage-time integrals.55
Sinus Node Recovery: is it Influenced by the Duration of AF, Reversible with Arrhythmia Termination, or Doomed from the Start of Arrythmia?
While electrical remodeling has been shown to occur in both CAF and PAF, the SN has been studied for its propensity for reverse electrical remodeling. The duration of AF is an important predictor of the restoration of sinus rhythm by direct cardioversion.56 Differences between SN electrical remodeling in chronic versus paroxysmal atrial fibrillation have been studied. In one paper, comparing patients undergoing left atrial catheter ablation for CAF and PAF, CAF was associated with slower reverse electrical remodeling.42 Patients with PAF had shorter AERPs following AF termination as compared to baseline. In this patient group AERP returned to baseline within five minutes. In patients with CAF, both AERP’s and SNRT’s were shortened, but it took up to three weeks to return to baseline duration.42 Another study by Raitt et al. noted that AERP varies significantly at the lateral right atrium, as compared to the proximal and distal coronary sinus.57 They concluded that electrical remodeling occurs at different rates in different regions of the atrium, which could be a possible trigger for future atrial arrhythmia.57
WAforementioned studies on canine atrial memory also noted differential findings between the left and right atria and demonstrated “memory” of atrial arrhythmia in the left atrium only. Also, it may be interpreted that the SN may be more likely to recover, as a RA structure, with less atrial memory/remodeling present than the LA. These findings may also explain why the left atrium is the major focus of catheter ablation in patients with recurrent AF. In a study by Hocini et al twenty patients with PAF and prolonged sinus pauses ( >3 seconds) were ablated and resulted in significant reverse remodeling of SN function with increase in heart rate, heart rate range, maximal heart rate and decreased SNRT.20 The extent of damage to the SN, whether from ischemia or chronic atrial arrhythmia may determine its reversibility of function. A study by Waris et al. showed that if the SN has sustained only partial damage to its structure, it is more likely to recover after DC cardioversion even after years of AF.58
In another study by Daoud et al., progressive recovery of SNRT and P wave amplitude T were observed in patients with chronic AF only after 14 days and much more after 3 months post ablation.47 For some individuals, duration of AF/AFL may not be critical: in a case report by Palma et al. SN function was noted to return to normal one day following ablation of chronic (25 years) atrial flutter.22 Finally, recovery of SN function has been demonstrated in patients with CAF following the surgical maze procedure, where SND disappeared after 12 months59 and SN response to exercise returned after 6 months after Maze procedure.60 The recovery of SN function again raises “chicken or the egg” questions. Is the SN more likely to recover if it was not damaged prior to the arrhythmia? Given that some AF terminations reveal SN dysfunction, can it be that AF developed as a “rescue” mechanism to SND and by terminating the AF, the underlying SN disease is now uncovered? We have seen that SN function normalizes in some patients after years of AF, so AF does not always cause lasting damage to the SN. Yet in others atrial enlargement in CAF causes irreversible structural changes to the SN.
Studies of electrical remodeling and cardiac memory help to elucidate the relationship between AF and SSS. However more information is required to understand the link between these two entities. Evidence reviewed above supports the notion that AF results in SN remodeling on a cellular and molecular basis and may cause SND.23,25,32,36,40,41 However not all patients with AF have SSS, and many present with SSS prior to developing AF. Animal models of SSS clearly demonstrate that SND can presage the onset of AF.61 Ultimately, factors common to both conditions such as aging and interstitial atrial fibrosis, may explain their coexistence.
Treatment of patients presenting with both AF and SSS (tachycardia-bradycardia syndrome) is particularly challenging. One can divide patients into three main groups: 1) patients with AF and reversible SND; 2) patients with AF and permanent SND requiring pacemaker implantation, and 3) patients with AF who have reversible SND but are not candidates for rhythm control therapy. Patients with permanent SSS will clearly benefit from pacemaker (PPM) implantation and medical therapy. The difficult task will be predicting which patients have “permanent” SSS following eradication of AF.
Our review raises many unanswered questions that relate to patient management.
1.Sinus node dysfunction in atrial fibrillation: cause or effect? Chicken or egg? Can be both or either.
2.Could it be that SND gives rise to PAF, and then via process of electrical remodeling or atrial memory, PAF promotes itself to CAF and in turn worsens the SN function? AF begets AF? Yes!
3.Could the two processes be coexistent, since many of the structural abnormalities and etiologies of both are similar? Yes!
4.Could AF be the rescue mechanism of SN after all? Yes!
5.Can management of one worsen the other? (maze, ablations that worsen SN function, antiarrhythmic medications, and pacemakers that may induce AF?). Yes!






