Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia that contributes to mortality.1 AF increases mortality risk2 and increased morbidity due to increased risk of stroke, heart failure and myocardial infarction.3-5 Many factors predispose patients to the development of AF, such as surgery,6 hypertension, valve disease, coronary artery disease or heart failure; however the strongest predisposing factor is ischemia.7 Yet, understanding risk factors has not improved patient survival. Current therapies for AF are suboptimal and their potential pro-arrhythmic effects limit their benefit.8 Research has focused on understanding the mechanisms underlying AF onset and maintenance in the hope of developing more targeted beneficial treatments. Research has begun to focus on the importance of slowed electrical conduction through atrial tissues in the development and maintenance of AF. Gap junctions, made up of connexins, are principle components responsible for both atrial and ventricular myocardial conduction. In this review, we will focuses on the relationship between connexins and mechanisms responsible for the development and maintenance of AF.
A summary of the current understanding of AF is that initiation occurs through either reentry or focal ectopic firing. In lone AF, continued ectopic firing from pulmonary veins or other sites may play a role in sustaining the arrhythmia as well. In the setting of structural heart disease, most studies suggest that AF is sustained through intra-atrial reentry.9,10
Focal ectopic firing that initiates, and in some circumstances sustains, AF can manifest as early-afterdepolarizations (EADs) or delayed afterdepolarizations (DADs). EADs can occur with action potential duration (APD) prolongation, which allows sodium or L-type Ca2+ channels to recover from inactivation to depolarize the membrane. This mechanism may explain the increased prevalence of AF in some long-QT patients.11 Studies have also shown involvement of the sodium-calcium exchanger in the development of either EADs or DADs.12 DADs occur when abnormal Ca2+ release during diastole causes an inward current by activating the Na+-Ca2+ exchanger, depolarizing the membrane. Several reviews have been written on these topics and other ion channel changes, such as increased K+ currents, that occur in the setting of AF.13,14
Reentry requires tissue heterogeneities in conduction and repolarization. An important contributor to these elements are atrial structural and electrical remodeling that occur with sustained AF. The phrase ‘AF begets AF’ summarizes how these remodeling events resulting from AF increase the probability of reentry and thereby increase the probability of further AF. Electrical remodeling refers
to alterations in ion channels that promote the development of AF. Examples include up-regulation of IK1 and IKACH, which lead to APD shortening.15,16 Similarly, Ca-channel down-regulation and changes in calcium handling in AF also lead to APD shortening. Most important in structural remodeling is the development of atrial fibrosis, which can interfere with electrical coupling and cause conduction slowing.17 Both APD shortening and CV slowing are inherent mechanisms in sustaining AF. Wavelength theory states that a reentrant wave is dependent upon conduction velocity (CV)and refractory period. Both decreased CV and shortened APD occur in AF, which leads to maintenance of AF. Drug-induced or gene-therapy induced APD prolongation has proven effective in suppressing AF.8,19 Recently, we have also shown that enhancing CV through overexpression of connexins 40 or 43 (Cx40, Cx43) reduces the incidence of AF.10 This highlights the importance of connexin remodeling in the setting of AF.
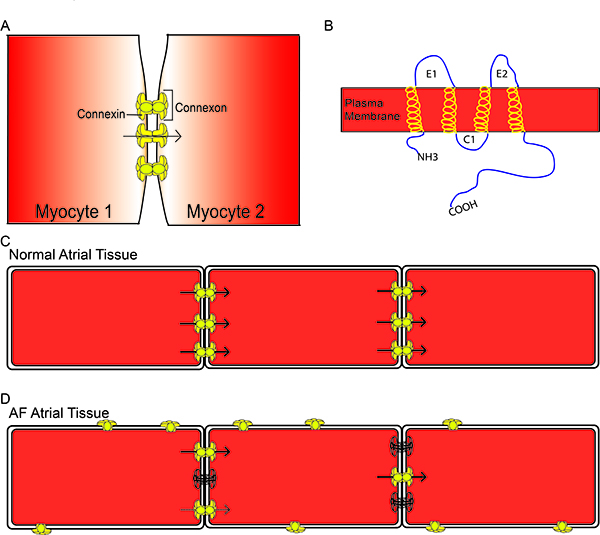
Connexins are ubiquitous proteins that can be found in most organs including atrial and ventricular myocardium and the specialized cardiac conduction system. Each connexin has a general structure of 4 transmembrane helices, 2 extracellular loops, a cytosolic loop and cytosolic N- and C-termini (Figure 1B). Six connexins join together to form a hemichannel, which docks with a hemichannel on an adjacent cell to form a gap junction (Figure 1A). Gap junctions are large conductance channels between adjacent cells that allow for low resistance passage of metabolic substrates and ions from one cell to another (arrow Figure 1C). In the heart, intercellular connectivity through gap junctions is a fundamental element controlling conduction of the electrical impulse across the myocardium. Atrial fibrillation, heart-failure, myocardial ischemia and infarction have all been associated with heterogeneous or decreased expression of Cx43 and decreased CV, suggesting a tight association between connexins and conduction.20,21 Several isoforms of connexins exist and they are named according to their molecular weight. In the atrial myocardium, Cx40 is the predominant connexin followed by Cx43 and Cx45.22 Studies have found strong involvement of Cxs 40 and 43 in the development and maintenance of AF but little is reported on Cx45 in AF.
Connexins are highly regulated proteins such that they respond to intracellular voltage changes or acidification, and their function is altered by phosphorylation.23-26 Alterations in connexin phosphorylation status lead to changes in connexin localization and/ or conductance. Connexins are normally expressed at the polar ends of myocytes in the intercalated disks (Figure 1C). Under stressed conditions such as ischemia or AF, connexins undergo changes in phosphorylation that ultimately lead to changes in connexin localization away from the intercalated disks and changes in gap junctional conductance for the connexins remaining at the disks (Figure 1D).
Figure 1. General Connexin Structure Six individual connexins form a connexon which docks with a second connexon, forming a gap junction (A). Connexins have the general structure of 4 transmembrane -helices, 2 extracellular loops, a cytosolic loop, and a cytosolic N- and C- terminus (B). Connexins are normally located at the polar ends of myocytes in the intercalated disk and allow for the passage of metabolic substances and ions from one myocyte to the next (arrows) (C). In atrial fibrillation, there is a reduction in connexins at the intercalated disk (outlined connexins) and enhanced lateralization of connexins. There can also be alterations in phosphorylation of connexins, resulting in reduced conductance (dashed arrow) (D).
Connexins Involvement in AF
Human genetics studies point to a role for Cx40 in the initiation and maintenance of AF. Although the majority of AF is thought to be sporadic and non-familial, connexin mutations have clearly been associated with familial, early onset AF in patients as summarized in Table 1. Human studies have identified Cx40 nonsense mutations, gene promoter polymorphisms and somatic mutations that lead to an overall reduction in Cx40 expression.27-30
Although Cx40 genetic mutations have been identified in the development of AF, public screening for Cx40 mutations to explain lone AF would not be cost-effective. Evidence suggests family history of AF increases the risk of development of AF31,32 but Cx40 disease-causing mutations have been infrequent and polymorphisms that change neither Cx40 expression nor function have also been identified.33 Screening for AF risk genes may prove beneficial in the future, if more Cx40 mutations become linked to development of AF. Currently, Cx40 gene mutations have been identified in only several cases of familial AF.
Although Cx40 genetic mutations have not been identified as the major cause of familial AF, altered Cx40 expression has clearly been observed in patients with AF. Both genetic mutations in Cx40 and non-genetic based AF have led to connexin trafficking defects,reductions in gap junctional conductance and altered expression of Cx40. Nao et al found AF patients with mitral valve regurgitation had heterogeneous reductions in Cx40 expression.34 In contrast, Polontchouk et al and others found AF patients expressed higher levels of Cx40, but they found increased connexin lateralization (Table 1).35,36 Although changes in total Cx40 expression differ between studies, common findings between these human AF studies include heterogeneous expression of Cx40 across different regions of the atria and lateralization of Cx40 throughout the atria, both of which could lead to heterogeneous and slow conduction and potentially to reentry.
Table 1. Connexin 40 in Human AF: Examples of studied Cx40 genetic mutations found in AF patients and findings of Cx40 alterations in patients with AF.
| Connexin 40 in Human AF |
|---|
| Gene Mutations | Effect on protein expression | AF effect | |
| Nonsense mutations Cx40 promoter SNPs | Stop codon inserted resulting in truncated Cx40 at amino acid 49 -44 Glycine Alanine | Familial AF with 100% Penetrance | Yang et al 2010 |
| | Increased atrial vulnerability (i.e. easy | Firouzi et al 2004 |
| | Early onset AF | Wirka et al 2011 |
| Missense Mutations | TATA box SNP (rs10465885) resulted in decreased Cx40 mRNA expression Proline 88 Serine or Glycine 38Aspartate, resulting in no Cx40 at the intercalated disc Alanine 96Serine resulting in low conductance GJ Methionine 163Valine, no GJ phenotype observed | All observed in early onset AF | Gollob et al 2006 |
| Patients in AF With mitral valve disease | Increased Expression of Cx40 but with increased lateralized Cx40 and Cx43 Increased Cx40 expression | Study of 12 chronic AF patients | Polontchouk et al 2001 |
| Reduced and heterogeneously expressed Cx40 | Study of 41 lone AF and 36 AF patients with valve disease | Wetzel et al 2005 |
| | Study of 10 patients with valve disease and AF | Nao et al 2003 |
Several animal models have been developed to study AF, including atrial burst pacing with and without heart failure, and ventricular failure with increased AF inducibility but without sustained AF. Similar to the human data, a common finding across these diverse models has been heterogeneous expression of Cx40. Further supporting a role of Cx40 in AF, Cx40 knock-out mice have abnormal atrial impulse propagation, slowed conduction, decreased safety factor for 1:1 conduction in the atrium and increased arrhythmia inducibility.37-40 Contrary to adult mice studies, in vitro cultured strands of neonatal atrial myocytes from Cx40 knock-out mice showed enhanced conduction velocity with an associated increase in Cx43 expression at the intercalated. A confounding factor for these cell culture studies was the finding of increased Cx43 in the atrial myocytes isolated from the Cx40 knock-out neonates. The increased conduction velocity may simply be a function of Cx43 levels in that experimental system.
While there may be some differences in findings between the various experimental models (Table 2), the majority of animal evidence supports alterations in Cx40 expression and/or localization, associated with reduced CV and increased AF vulnerability. These studies corroborate the human data strongly suggesting an integral role of Cx40 in AF.
Table 2. Connexin 40 in animal models of AF
| Model | Effect | |
|---|
| Cx40 KO mice | Atrioventricular conduction slows | Simon et al 1998 |
| Atrial Conduction slowing, with high frequency of intra-atrial reentrant tachycardia | Kirchhoff et al 1998 |
| Altered atrial conduction patterns, decreased right atrial conduction and decreased safety factor for maintaining 1:1 conduction | Bagwe et al 2005 |
| Loss of right atrial and left atrial CV heterogeneity, Cx40 developmentally required for setting up heterogeneity | |
| Cx40 KO neonatal cardiomyocytes | Increased propagation velocity in synthetic strands of Cx40 KO cardiomyocytes. This is associated with increased Cx43 in the intercalated disc. | Beauchamp et al 2006 |
| Rapid-pacing induced AF in goat | Total Cx40 expression and CV is unchanged, but heterogeneous expression of Cx40 | Van der Velden et al 1998 |
| Cx40/Cx43 ratio decreases as increased stability of AF | Van der Velden et al 2000 |
| Rapid-pacing induced AF in pig | Total Cx40 expression same as sinus-rhythm; however, intra-atrialconduction reduced | Igarashi et al 2012 |
Similar to Cx40, inconsistencies in Cx43 expression between the various studies have been shown (Table 3). Rare familial AFassociated Cx43 mutations have been found that alter connexin trafficking, resulting in a mosaic pattern of connexin expression across the atrium, suggesting that decreases in Cx43 can cause AF.41 Wetzel et al. found that AF patients with mitral valve disease had increased Cx43 expression.35,36 Rucker-Martin found no change in overall Cx43 expression but relative increased in the amount of unphosphorylated and/or lateralized Cx43. 42 Importantly, studies have found that total Cx43 expression decreases in heart failure, but the AF studies have generally not had a significant component of heart failure patients in their populations. Overall, the human data on Cx43 is less robust than the data on Cx40, but the available data suggest that alterations in Cx43 expression or function may cause AF, and that AF may reduce the amount of functional, intercalated disklocalized Cx43 if not the total Cx43 amount.
Table 3. Connexin 43 in Human AF: This table lists examples of both genetic mutations and observed Cx43 alterations in patients with AF.
| Gene Mutations | Effect | | |
|---|
| Frameshift Mutation | Nucleotide Deletion resulting in intercellular retention of GJs and dominant-negative down-regulation of both Cx40 and Cx43 | Sporadic nonfamilial case of AF | Thibodeau et al 2010 |
| Patients with AF With mitral valve disease | Increased Cx43 expression | Study of 36 AF patients | Wetzel et al 2005 |
| With dilated atria | Both dilated atria and AF patients had dephosphorylated and lateralized Cx43. | Study of 5 AF patients and 5 patients with dilated atria | Rucker-Martin et al 2006 |
Animal models of AF more strongly support the involvement of Cx43 in AF than the available human data. Several animal models show alterations in Cx43 expression associated with AF (Table 4). In the neonatal mouse in vitro myocyte culture system described above, Beauchamp found atrial strands from Cx43 knock-out mice have shown reduced CV and the substrate for reentry, but their Cx43 findings were confounded by the finding of decreased Cx40 expression in the myocytes isolated from the Cx43 knock-out mice.43 Further highlighting the importance of Cx43 in atrial conduction, transgenic mice carrying the Cx43 G60S mutant (described in occulodentodigital dysplasia) are highly susceptible to induction of atrial tachycardia and atrial arrhythmias.44 Similarly, a rapid pacing pig model of AF and severe heart failure had decreased Cx43 expression, decreased phosphorylated Cx43, enhanced lateralized connexins (Figure 2) with CV slowing and persistent AF.10 In addition gene-therapy expressing Cx43 preserved atrial conduction and prevented the development of AF, further supporting Cx43 as a major contribution to atrial stability.10
Figure 2. Atrial staining for Connexin 40 and 43 Rapid-pacing induced AF in a porcine model showed no alteration in Cx40 expression (yellow); however, there was reduced expression of total and phosphorylated Cx43 and enhanced lateralization of Cx43 (green). This resulted in significantly less Cx43 localized in the intercalated disc (IC-disk), as shown in A with quantification in B. IC-disk was calculated by (IC-disk intensity/total intensity) x 100% Adapted from Igarashi et al 2012

Table 4. Connexin 43 in Animal Models of AF
| Model | Effect | |
|---|
| Congestive Heart Failure canine | Decrease in phosphorylated Cx43 associated with an increase in lateralized Cx43. Also interstitial fibrosis present, leading to heterogeneous conduction in atria. | Burstein et al 2009 |
| Rapid-pacing induced AF in goat | Total levels of Cx43 and localization unchanged, increase in dephosphorylated Cx43. | Van der Velden et al 1998, 2000 |
| Rapid-pacing induced AF in pig | Reduced and lateralized expression of Cx43. | Igarashi et al 2012 |
| Point-mutation Cx43 G60S mutant (Oculodentodigital dysplasia mouse) | Heterozygous mutant mouse, dominant negative decrease in total Cx43 expression. Results in a 50% reduction of myocyte junctional conductance, highly susceptible to induction of AF/sustained AF. | Tuomi et al 2011 |
| Cx43 KO embryonic cardiomyocytes | No expression of Cx43 but also a decrease in Cx40 expression. Decrease in conduction velocity. | Beauchamp et al 2005 |
| Cx43 heterozygous KO mice | 50% reduction in atrial Cx43 expression, but no change in atrial CV | Thomas et al 1998 |
| Sterile pericarditis in canine | Overall reduction in Cx43 expression, CV slowed and increased sustained atrial arrhythmias (AF or atrial flutter) | Ryu et al 2006 |
Although strong evidence supports Cx43 involvement in determining atrial conduction properties and potentially increasing vulnerability to development or maintenance of AF, not all studies have shown this. Thomas et al reported no changes in atrial CV or P-wave duration in Cx43 heterozygous knock-out mice.45 Overall, the data on Cx43 show association of Cx43 mutations with AF in very rare cases, correlation between Cx43 expression and intercalated disk localization and atrial conduction properties in most but not all studies, and a reduction in Cx43 expression resulting from AF in the setting of heart failure but not necessarily in the setting of normal ventricular function.
Therapies to terminate or prevent AF include antiarrhythmic drugs, the surgical maze procedure, and ablation either to isolate pulmonary veins or to disrupt intra-atrial reentry. Pulmonary vein isolation is successful in terminating AF in up to 80% of patients, with greater success occurring in patients with paroxysmal, lone AF.47 Ablation of reentrant circuits is considerably less successful, with long-term freedom from AF less than 50% in patients with dilated atria and persistent or permanent AF.49 Limitations to ablation success include inadequacies in currently available tools to create complete lines of conduction block and the lack of a generally accepted lesion set based on the underlying mechanisms responsible for maintaining AF.
Better atrial imaging and mapping techniques are being developed to improve ablation efficacy. MRI imaging has successfully identified gaps through scarred atrial regions which provide the site of reentry for AF, allowing for more targeted ablations.50,51 In contrast, another more recent study did not identify visible reentrant pathways on MRI even though reentrant circuits were found within the scarred regions.52 Similarly, Focal Impulse and Rotor Modulation (FIRM) techniques to identify rotors and focal sources of AF show promise in creating smaller ablation lesions while still providing AF termination.53,54 In canine models, extensive ablation of both the right and left atrium was required to get 100% reduction of AF. However this was associated with decreased Cx43 expression in areas near the ablation site.55 This finding suggests that less extensive ablation may have the unintended consequence of worsening conduction heterogeneity, which may be proarrhythmic.
Research has focused on developing therapies that target both connexin and ion channel remodeling, in hopes of altering the underlying mechanism thereby preventing development of sustained AF. In canines, bepridil was found to reduce AF inducibility and duration, while simultaneously preventing SCN5A and L-type Ca2+ channel reductions.56 Pairing bepridil with olmesartan also prevented Cx43 down-regulation and reduced tissue fibrosis, both resulting in improved conduction velocity.57 This bepridil/olmesartan strategy successfully prevented AF inducibility, but results of this strategy for patients with already remodeled atria are unclear. A similar study using amiodarone showed reverse-remodeling, which may be a more relevant end-point.58 Of course, many patients with AF are already treated with amiodarone. Since this drug is not curative, more extensive investigation is needed to develop a definitive therapy. Recently relaxin, an anti-fibrotic hormone, showed promising atrial fibrosis reverse-remodeling in spontaneous hypertensive rats; however, large animal studies need to be done to evaluate translational possibilities.59 Similarly, metoprolol was found to antagonize connexin lateralization and conduction slowing in AF,60 however, metoprolol also is not curative for AF. These drug data emphasize the complexity of the situation and the need for extensive analysis in realistic test models.
Targeted connexin therapies are in early stages of development. We explored the possibility that targeted overexpression of either connexin 40 or 43 in the atrium would improve CV and prevent AF in our porcine AF-heart failure model.10,61 The model combined 42-hz burst atrial pacing used in the goat lone AF model with an uncontrolled ventricular response giving tachycardia-induced ventricular failure. We delivered the gene for either Cx40 or Cx43 using an epicardial painting method that we had previous validated to delivery genes densely and homogeneously to the atria.62 In sinus rhythm animals, we saw no change in the already normal CV with either Cx40 or Cx43 overexpression. Since connexin expression and post-translational processing were normal, we interpreted these data to suggest that gap junctional conductance was not the limiting factor to CV under normal circumstances. In the AF-heart failure animals, we saw normalization of CV and prevention of AF with gene transfer of either connexin. These data linked connexin expression to CV alterations in AF and demonstrated that CV played a role in AF development and maintenance. Bikou et al. found similar results with Cx43 overexpression using a more localized inject and shock delivery method in the pig AF-heart failure model.61
Drug therapies targeting connexins have also been found to
enhance atrial CV.63 Rotigaptide is a small peptide that has been
show to antagonize CV slowing in a variety of situations. Shiroshita-
Takishita et al. found improved conduction in 3 canine models of
atrial remodeling; atrial tachypacing, ventricular tachypacing and
acute atrial ischemia, but AF vulnerability was reduced only in acute
ischemia.64 Similarly, Haugan et al showed that rotigaptide enhanced
atrial CV but did not reduce AF inducibility in a rabbit model of
chronic volume overload.65 Similarly, Guerra et al. found rotigaptide effective in prevention of AF in mitral valve regurgitation but not
congestive heart failure.66 These studies suggest that interaction
between connexin expression and function and AF development may
vary by model. A limitation of this conclusion is that rotigaptide’s
mechanism of action is not fully understood. Further studies need
to be performed to understand the possibilities for gap junctiontargeted
therapies for either prevention or treatment of AF.
Connexins are highly dynamic proteins that are important in myocardial conduction. Overall, inconsistencies exist over the exact pattern of connexin expression changes in the setting of AF. Although less common, genetic mutations in Cx40 and Cx43 have been found as the definitive cause of AF in both human studies and animal models. Importantly, both animal studies and human studies of AF commonly show connexin phosphorylation is altered, connexins become lateralized and connexins are heterogeneously expressed. Alterations in connexin phosphorylation and localization have been associated with decreased atrial conduction. Heterogeneous and slowed atrial conduction creates the substrate conducive to reentry. Taken together, connexin remodeling is a key mechanism in the maintenance of sustained AF. Several drug-therapies, genetherapies and small peptides targeting connexin remodeling have proven beneficial in antagonizing connexin dephosphorylation and down-regulation in AF, leading to enhanced CV and reduced AF vulnerability, verifying the importance of connexins in the initiation and maintenance of AF. Future studies need to be done to assess the potential role of these therapies in the setting of atrial fibrosis, structural remodeling or pre-existing AF.






